I’m in Thailand and I did no blood test. I acted on a theory.
From what I can tell, Sulforaphane boosts mind clarity and focus but depletes L-Tyrosine, Dopamine and Serotonin.
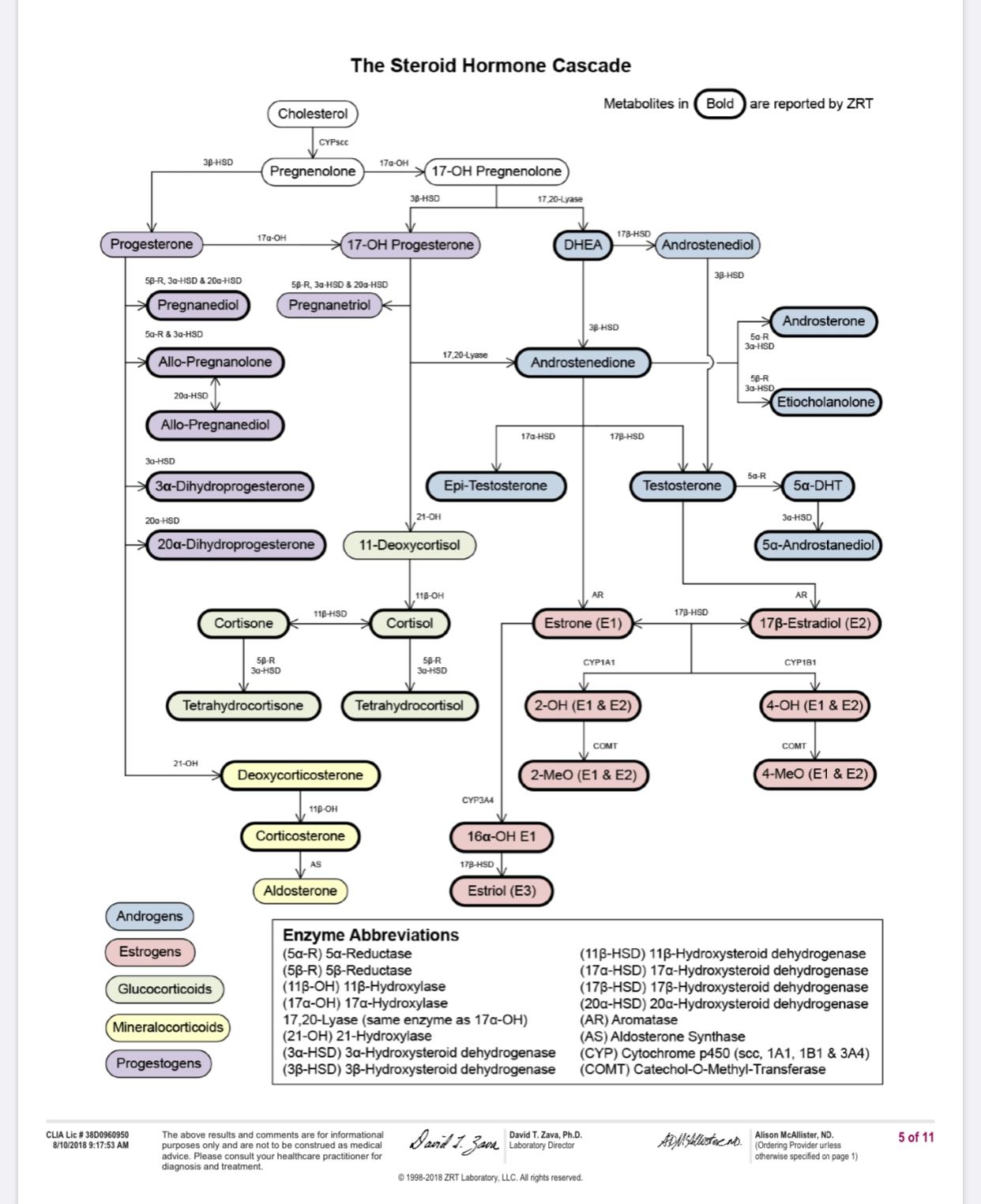
I have no evidence that it did anything to my 3a-HSD, or if it did, that 3a-HSD improved my insomnia or stress levels. 3a-HSD + 5-AR + Progesterone = Allopregnanolone which should regulate GABA a receptors, fix insomnia and a whole bunch of other things.
However, that is if every part is doing its work correctly which I now doubt. Some parts seems to have gotten long term damage. What works best for me is very low carbs diet, with lots of saturated fat and some mono unsaturated, and lots of fat cuts of meat. Then some exercise and weight lifting on top on this. I believe this is repairing the long term damage as I have seen improvements on every symptoms. Most of them are gone actually. (after 13 months of diet)
No easy button for this one. I don’t think a pill will do the trick. I had to take the hard path.
I attribute it to having more Beta-HydroxyButyric acid in my blood by doing a ketogenic / carnivore diet.
It lowers Glutamate (excitatory), rises GABA (calming) and some research indicates it may repair damage done to the DNA although I can’t say if it’s true or not.
I don’t know if it influences Allo. I looked at 5a-DHP and it seems to work but it’s dose dependent.
If you do try it, please let us know the results !
My latest research and what I am looking into involves the enzymes 3a-hydroxysteriod and 3B-hydroxysteriod. I forgot that we already started going down this path until I found this thread again. @Dubya_B I agree with what you and I also believe that Fin and Dut can effect the 3a-HSD and 3b-HSD enzymes by binding NADPH. I also believe that the way Fin and Dut effect 5AR is by binding NADPH. We have been led to believe for many years that 5AR inhibitors “inhibit or decrease the actual 5AR enzyme” I don’t believe that this is correct. Instead I think Fin and Dut temporarily inhibit T-DHT conversion by preventing the 5AR enzyme from being able to use it’s cofactor NADPH to catalyze the conversion of T-DHT. I believe that the main difference between people who develop PFS and people who don’t comes down to issues with binding NADPH and that any enzyme that needs NADPH to catalyze it’s function has the potential to be effected.
“Mechanism-based inactivation of 5α-reductase type 2 by finasteride showing a hydride reaction occurring between finasteride and NADPH that results in a covalent bond between finasteride and the cofactor. [R = -C(=O)-NH2; PADPR = 2’-phosphoadenosine-5’’-diphosphoribose; A-H = TYR58.7] Finasteride in relation to NADPH while bound to 5-beta-reductase, note the distance between the hydride on NADPH (purple) and the sp2 hybridized carbon on finasteride (green)”
“Finasteride is a selective 5α-reductase type 2 inhibitor that reduces plasma 5α-dihydrotestosterone levels and shrinks the size of the prostate (7). … Subsequent to inhibitor binding*, there is hydride transfer from the NADPH cofactor to the Δ1-2-ene double bond of finasteride”
“Testosterone is irreversibly converted in a nicotinamide adenine dinucleotide phosphate (NADPH)-dependent reduction reaction to the most physiologically active androgen 5α-dihydrotestosterone (DHT)”
“Circulating testosterone diffuses into prostatic cells, where it is irreversibly converted into the more potent androgen DHT via the NADPH-dependent reduction catalyzed by the steroid 5α-reductase type II and, to a lesser extent, by steroid 5α-reductase”
“finasteride (Proscar®; Propecia® [Merck, NJ, USA] Figure 1), which was approved for benign prostatic hyperplasia (BPH) and alopecia, respectively [203], and the dual steroid 5α-reductase (type I and II) inhibitor dutasteride (Avodart® [GlaxoSmithKline, UK]), approved for BPH [204], mediate their effects by mechanism-based inhibition of steroid 5α-reductase type II”
“The mechanism of 5α reductase inhibition is complex, but involves the binding of NADPH to the enzyme followed by the substrate”
So clearly Fin and Dut are effecting NADPH. They are not just “inhibiting the 5AR enzyme”. With this being said it makes since that every enzyme that need NADPH as a cofactor will be effected
I would also like to see some studies that support the idea that 3a-HSD and 5AR are “mutual antagonists”. This theory is interesting.
I tried 5a-DHP. Others have as well. It’s certainly dose dependent and it’s literally putting a downstream neurosteroid in your body which is uncharted territory and risk with an unpredictable result. But like many others I have decided that taking risks is worth the possibility of fixing this.
The main thing about 5α-Dihydroprogesterone (5a-DHP) is that it converts to allopregnanolone and back to 5a-DHP via 3α-Hydroxysteroid dehydrogenase.
3a-Hydroxysteroid dehydrogenase is the enzyme that we are discussing in this thread as an enzyme that at least some PFS people may have an issue with. So seeing that 3a-Hydroxysteroid dehydrogenase converts 5a-DHP into allopregnanolone and allopregnanolone back to 5a-DHP it appears to be a “regulatory” function. Meaning if we need more allopregnanolone this enzyme can give us more from 5a-DHP. And if we need less allopregnanolone and more 5a-DHP it can convert allopregnanolone back into 5a-DHP. So if we have an issue with 3a-Hydroxysteroid dehydrogenase this “regulatory” process can be disrupted and the result can be unpredictable. Also if we are short in allopregnanolone because we have an issue with 3a-Hydroxysteroid dehydrogenase not being able to convert 5a-DHP into allopregnanolone than taking more 5a-DHP may not help. Because we could be giving our system more 5a-DHP that can not convert into allopregnanolone. So I think the best way to go about this is test for allopregnanolone which I am in the progress of doing and if my allopregnanolone is low take allopregnanolone directly.
I agree. I theorize that the only thing we all have in common is that we took the same drug which effected NADPH, had the same/similar problems/symptoms. However, I think there are several different enzyme disorders and imbalances that a person can end up suffering from who have a predisposition to having issues in result to taking drugs that bind NADPH. Upon doing some research it becomes clear that a lot of these enzymes use NADPH as a cofactor and that’s what we have in common. But the exact enzyme disorder and imbalance that resulted differs from person to person.
I agree serum DHT levels although we need to know what they are for our own cases they don’t seem to have much significance as far as giving us clues as to the mechanism behind our disorder. Our serum DHT levels clearly do now show any issues or at least major consistent issues that would explain our side effects. However, normal or high serum DHT levels do not necessarily correlate with what our peripheral genital skin DHT levels are. I speculate that some would probable find low peripheral DHT skin issues but it would not be a finding in all of us that would explain all of our symptoms. The studies found altered neurosteroids in PFS victims when compared to controls and I think that’s the clue that has been overlooked. If we all had access to downstream neurosteriod metabolite testing we could find our individual problem as well as determine which enzymes were effected by Fin/Dut binding NADPH.
Obviously the golden question is why after Fin/Dut left our system and NADPH was no longer bound why could the NADPH dependent enzymes not start doing their thing again and getting everything back in balance? Of course even if they could I’m sure different receptors up regulated and down regulated attempting to correct the imbalance. But in theory if we could correct the imbalance the receptors should go back to normal as well.
Never intended that interpretation of anything I’ve written, …unless it was sarcasm. I’ve only pointed out that the molar concentration of fin/dut (and 5-AR enzymes) is likely to be entirely insignificant compared to the concentration of NaDP+/NaDPH in any given cell. This was to debunk an absurd theory that was floating around here a couple years ago.
Molar concentration would not be so relevant if fin/dut act as catalysts in disrupting the natural processes and are not used in the reaction itself. Nonetheless, fin/dut is now absent, in my case for 2 years now, and I still have the imbalances so the damage is elsewhere. Considering the fluctuation of the symptoms, their appearances / dis-appearances, I would think of fluctuations in the epigenome as it still tries to find a balance.
Fin/Dut are depleted after becoming part of the 5-ar+NaDP+Fin/Dut complex. By definition, this means they are not a catalyst, but a reactant, and are biochemically inactive (as far as forming new covalent bonds) after forming this complex.
I did take sulforaphane in order to boost 3a-HSD, knowing it would decrease my DHT. I took as a premise that since my T and DHT were exactly at normal middle of the range for my age, my 5ar must be working normally. But I do have Allopregnanolone and (Allo)tetrahydrodeoxycorticosterone problems, the two of them being major anxiolityc and GABA-A modulators, as I can sleep more than a certain numbers of hours and then I completely wake-up and spend the rest of the night tossing and turning. It’s my last major symptom I might add.
As you know, 5ar transform Progesterone into 5a-DHP which is then turned into ALLO by 3a-HSD. I can’t get ALLO but I do have 5ar, so I thought I must be missing progesterone or 3a-HSD. (and progesterone comes from pregnenolone transformed by 3b-HSD, which could also very well be missing)
As I noted in the thread ALLO enhancing drugs that might not have been considered, Each enzyme, neurotransmitter, hormone, neurosteroid etc… is balanced by an opposite one and they’re all interconnected. So if you boost one and only one, you counter it’s opposite and it unbalances the rest of them.
That’s what happened when I took sulforaphane. At first, I felt great: sharp mind, great motivation, lots of energy, but it seems I later depleted some other neurotransmitters or neurosteroids and I “crashed” (meaning I got worst than before for a time). I stopped sulforaphane and got back to baseline.
I will need further researches to really give you a satisfying answer. I didn’t consider fin/dut effects on NADPH. However, and I will explain later, I did tried Nicotinamide adenine dinucleotide (NAD) boosters in an effort to have NAD+ work in synergy with BetaHydroxy Butyrate (BHB) and the sirtuin proteins to re-balance the epigenome.
This is my whole purpose of carnivore / ketogenic diet, being cold/hot, 1 meal a day, being hungry, out of breath, exercising without glycogen etc… It activates the sirtuin proteins as it forces the body into survival mode without truly putting it at risk.
I believe our problem lies in our epigenome. Fin and or the withdrawal of fin, has forced our body to adapt our epigenome to match the circumstances. However, unlike DNA which by nature is a digital “memory” (in base 4, not binary), the epigenome is analogical and has no memory of its former states. So, IMO, I must together work on re-balancing my body while working on the sirtuin proteins which regulates epigenetic changes and the demethylase enzyme. This way, if I ever achieve a better balance, the sirtuins will have it marked on my epigenome.
If indeed the problem is epigenetic as I believe, I’m afraid we will never be the same as we were before, the epigenome having forgotten it. But we can be a different, better version than our pfs state and even maybe better than our pre-pfs state.
Ps:L-Glycine and properly dosed Gelsemine increases 3a-HSD, but 4ml of Gelsemine will kill you by paralysis. So good luck finding some. Fluoxetine (prozak) also increases 3a-HSD, or more to the point increases it’s efficientcy by a magnitude of 171.2. Other SSRI do the same but some people are here because of those so I think it’s pointless to try.
Then as you said, fin/dut would not be in large enough concentration to deplete NADPH levels, as those are at the center of many cell activities and are quite abundant.
Plus, we’re now living in the absence of fin/dut and the problems persist. The cause is elsewhere. Do you believe the problem is epigenetic ?
I do too. In my opinion, it affected more than just the epigenome of our AR receptors. Hormones, neurosteroids, neurotransmitters and even regular proteins are affected. And for that to happens, IMO, there has to be a problem in copying segments of our DNA, AKA problem on the epigenetic level.
@Dubya_B: Do you know anyone, or any protocole that fixed the pfs typical insomnia: sleeping just a few hours, then waking up fully awake all of a sudden and then turning and tossing the rest of the night, barely sleeping if you can call it sleep ?
Is the question does Fin/Dut become depleted after becoming part of the 5-ar+NaDP+Fin/Dut complex? (not that this is not an important question because it is)
OR is the question did Fin/Dut temporally becoming a part of the 5-ar+NaDP+Fin/Dut complex cause problems with 5AR and other enzymes?
I was not being sarcastic in my response above. I apologize if it came across that way. I’m not familiar with the theory that was floating around a couple of years ago and I’m not arguing against it or in favor of it simply because I don’t know what the theory is. Instead the point that I was attempting to make is that if the molar concentration of Fin/Dut (and 5-AR enzymes) is entirely insignificant compared to the concentration of NaDP+/NaDPH in any given cell why is it that when Fin/Dut becomes a part of the 5-ar+NaDP+Fin/Dut complex (be in temporally or permanently) do we experience such a large reduction in serum DHT concentrations? The point being if this is how Fin/Dut inhibits testosterone-DHT conversion by becoming a part of the 5-ar+NaDP+Fin/Dut complex it’s hard to agree that this is as insignificant as you suggest if its temporally inhibiting DHT to such a large degree.
Are you saying that Fin/Dut becomes a part of the 5-ar+NaDP+Fin/Dut complex and in addition works as a competitive inhibitor of the 5AR enzyme which explains the actual reduction of serum DHT levels while on the drug? If this is the case than why even make Fin/Dut a part of the 5-ar+NaDP+Fin/Dut complex? In other words why did the creators design the drug to become a part of the 5-ar+NaDP+Fin/Dut complex? You appear to have a background in biochemistry so you are probable a good person to ask. Is this the norm for inhibitor based drugs? To become a part of the cofactor? I have no background in this like most people here so all I can do is read on the internet. But upon doing so I cannot find another inhibitor based drug that inhibits it’s target enzyme by becoming a part of the enzymes cofactor. Am I wrong? I am totally open to the real possibility that I am wrong and hope that I am so I can rule this out and move onto the next.