How did you increase Allo? with 5a-DHP?
if we have an issue with 3a-HSD the 5a-DHP may have never converted into Allo. We need to get tested for Allo. This is the only way to know.

How did you increase Allo? with 5a-DHP?
if we have an issue with 3a-HSD the 5a-DHP may have never converted into Allo. We need to get tested for Allo. This is the only way to know.
do you still take Sulforaphane. The issue with every method your mentioned to increase Allo they require working 3a-HSD.
Never intended that interpretation of anything I’ve written, …unless it was sarcasm. I’ve only pointed out that the molar concentration of fin/dut (and 5-AR enzymes) is likely to be entirely insignificant compared to the concentration of NaDP+/NaDPH in any given cell. This was to debunk an absurd theory that was floating around here a couple years ago.
It’s very Ultimate!
Molar concentration would not be so relevant if fin/dut act as catalysts in disrupting the natural processes and are not used in the reaction itself. Nonetheless, fin/dut is now absent, in my case for 2 years now, and I still have the imbalances so the damage is elsewhere. Considering the fluctuation of the symptoms, their appearances / dis-appearances, I would think of fluctuations in the epigenome as it still tries to find a balance.
Fin/Dut are depleted after becoming part of the 5-ar+NaDP+Fin/Dut complex. By definition, this means they are not a catalyst, but a reactant, and are biochemically inactive (as far as forming new covalent bonds) after forming this complex.
I did take sulforaphane in order to boost 3a-HSD, knowing it would decrease my DHT. I took as a premise that since my T and DHT were exactly at normal middle of the range for my age, my 5ar must be working normally. But I do have Allopregnanolone and (Allo)tetrahydrodeoxycorticosterone problems, the two of them being major anxiolityc and GABA-A modulators, as I can sleep more than a certain numbers of hours and then I completely wake-up and spend the rest of the night tossing and turning. It’s my last major symptom I might add.
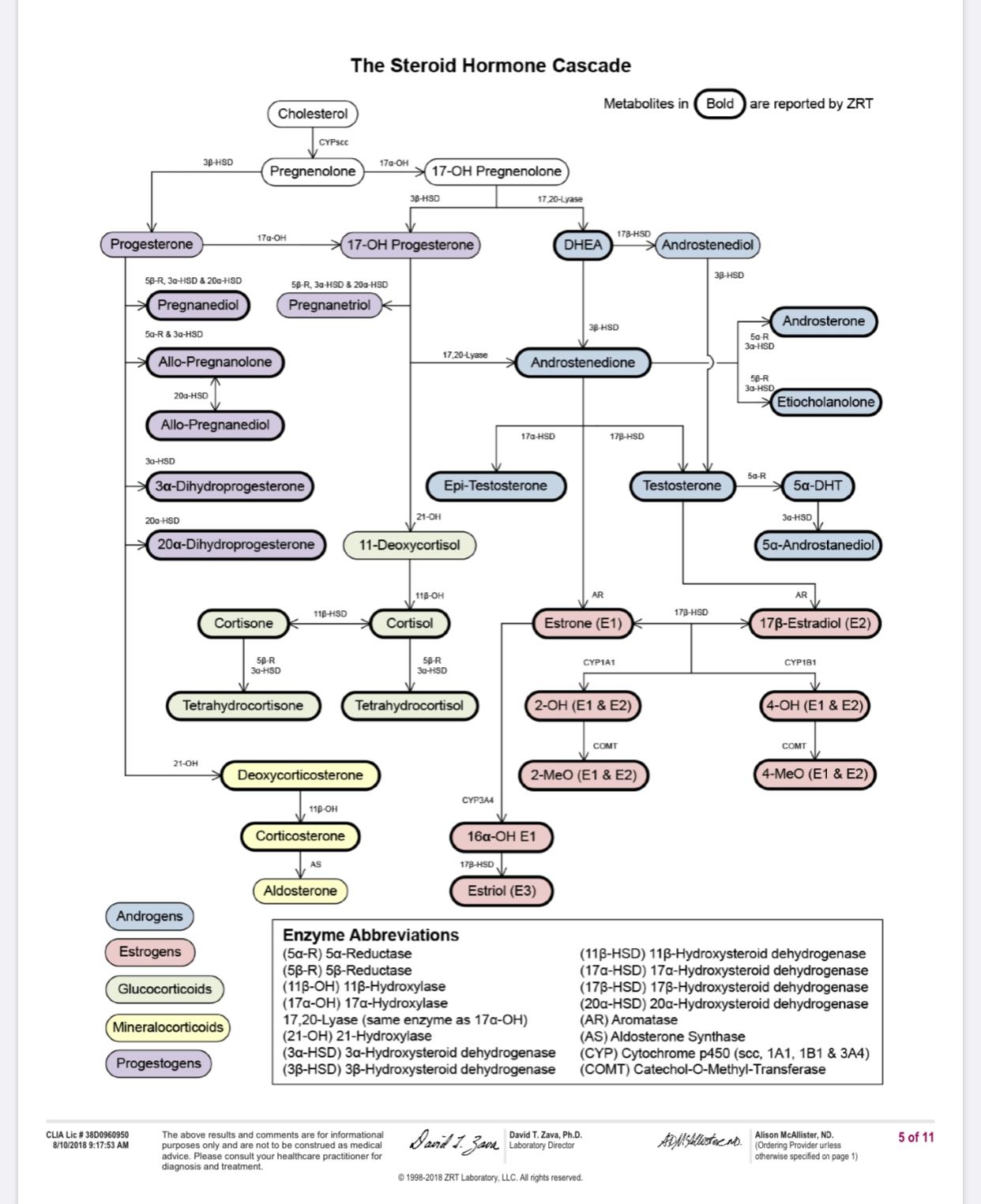
As you know, 5ar transform Progesterone into 5a-DHP which is then turned into ALLO by 3a-HSD. I can’t get ALLO but I do have 5ar, so I thought I must be missing progesterone or 3a-HSD. (and progesterone comes from pregnenolone transformed by 3b-HSD, which could also very well be missing)
As I noted in the thread ALLO enhancing drugs that might not have been considered, Each enzyme, neurotransmitter, hormone, neurosteroid etc… is balanced by an opposite one and they’re all interconnected. So if you boost one and only one, you counter it’s opposite and it unbalances the rest of them.
That’s what happened when I took sulforaphane. At first, I felt great: sharp mind, great motivation, lots of energy, but it seems I later depleted some other neurotransmitters or neurosteroids and I “crashed” (meaning I got worst than before for a time). I stopped sulforaphane and got back to baseline.
I will need further researches to really give you a satisfying answer. I didn’t consider fin/dut effects on NADPH. However, and I will explain later, I did tried Nicotinamide adenine dinucleotide (NAD) boosters in an effort to have NAD+ work in synergy with BetaHydroxy Butyrate (BHB) and the sirtuin proteins to re-balance the epigenome.
This is my whole purpose of carnivore / ketogenic diet, being cold/hot, 1 meal a day, being hungry, out of breath, exercising without glycogen etc… It activates the sirtuin proteins as it forces the body into survival mode without truly putting it at risk.
I believe our problem lies in our epigenome. Fin and or the withdrawal of fin, has forced our body to adapt our epigenome to match the circumstances. However, unlike DNA which by nature is a digital “memory” (in base 4, not binary), the epigenome is analogical and has no memory of its former states. So, IMO, I must together work on re-balancing my body while working on the sirtuin proteins which regulates epigenetic changes and the demethylase enzyme. This way, if I ever achieve a better balance, the sirtuins will have it marked on my epigenome.
If indeed the problem is epigenetic as I believe, I’m afraid we will never be the same as we were before, the epigenome having forgotten it. But we can be a different, better version than our pfs state and even maybe better than our pre-pfs state.
Ps:L-Glycine and properly dosed Gelsemine increases 3a-HSD, but 4ml of Gelsemine will kill you by paralysis. So good luck finding some. Fluoxetine (prozak) also increases 3a-HSD, or more to the point increases it’s efficientcy by a magnitude of 171.2. Other SSRI do the same but some people are here because of those so I think it’s pointless to try.
Then as you said, fin/dut would not be in large enough concentration to deplete NADPH levels, as those are at the center of many cell activities and are quite abundant.
Plus, we’re now living in the absence of fin/dut and the problems persist. The cause is elsewhere. Do you believe the problem is epigenetic ?
Yep.
I do too. In my opinion, it affected more than just the epigenome of our AR receptors. Hormones, neurosteroids, neurotransmitters and even regular proteins are affected. And for that to happens, IMO, there has to be a problem in copying segments of our DNA, AKA problem on the epigenetic level.
@Dubya_B: Do you know anyone, or any protocole that fixed the pfs typical insomnia: sleeping just a few hours, then waking up fully awake all of a sudden and then turning and tossing the rest of the night, barely sleeping if you can call it sleep ?
It’s a symptom I’d really like to tackle.
Is the question does Fin/Dut become depleted after becoming part of the 5-ar+NaDP+Fin/Dut complex? (not that this is not an important question because it is)
OR is the question did Fin/Dut temporally becoming a part of the 5-ar+NaDP+Fin/Dut complex cause problems with 5AR and other enzymes?
I was not being sarcastic in my response above. I apologize if it came across that way. I’m not familiar with the theory that was floating around a couple of years ago and I’m not arguing against it or in favor of it simply because I don’t know what the theory is. Instead the point that I was attempting to make is that if the molar concentration of Fin/Dut (and 5-AR enzymes) is entirely insignificant compared to the concentration of NaDP+/NaDPH in any given cell why is it that when Fin/Dut becomes a part of the 5-ar+NaDP+Fin/Dut complex (be in temporally or permanently) do we experience such a large reduction in serum DHT concentrations? The point being if this is how Fin/Dut inhibits testosterone-DHT conversion by becoming a part of the 5-ar+NaDP+Fin/Dut complex it’s hard to agree that this is as insignificant as you suggest if its temporally inhibiting DHT to such a large degree.
Are you saying that Fin/Dut becomes a part of the 5-ar+NaDP+Fin/Dut complex and in addition works as a competitive inhibitor of the 5AR enzyme which explains the actual reduction of serum DHT levels while on the drug? If this is the case than why even make Fin/Dut a part of the 5-ar+NaDP+Fin/Dut complex? In other words why did the creators design the drug to become a part of the 5-ar+NaDP+Fin/Dut complex? You appear to have a background in biochemistry so you are probable a good person to ask. Is this the norm for inhibitor based drugs? To become a part of the cofactor? I have no background in this like most people here so all I can do is read on the internet. But upon doing so I cannot find another inhibitor based drug that inhibits it’s target enzyme by becoming a part of the enzymes cofactor. Am I wrong? I am totally open to the real possibility that I am wrong and hope that I am so I can rule this out and move onto the next.
I suffered greatly in this department. In fact, my insomnia was so bad at one point that I stayed up for six weeks. I substantially improved my sleep issues with Glycine, magnesium and the herb ziziphus. However, I progress backwards fast if I stop taking these supplements. I could say that as long as I take these supplements my sleep is about 75 percent pre PFS. I don’t need ziziphus every night. But I need the Glycine and magnesium every night
unless we have a pre disposition to taking anything that binds to something that is at the center of many cell activities. This theory would certainly explain the variety of side effects.
a couple of practical but not so scientific points has me thinking along these lines. I took Dut for a month when I was 22 years old to treat an enlarged prostate. I had no issues on or after Dut. So if my issue is in result of having a genetic pre predisposition to experiencing epigenetic changes in result of taking a 5ar inhibitor my gut feeling tells me that this epigenetic change would have occurred immediately at the time of taking Dut how the syndrome manifest’s for everyone else while on or immediately after stopping the drug. Instead, my issues came four years later after taking saw P. That’s when I got PFS. Then three years after that I took Saw P again and got even worse this time completely crushed. So where I am going with this is that it appears for me this was an accumulative thing each time depleting something vital required for optimal functioning and that the difference between me and most PFS victims is that taking Dut once was not enough to deplete what ever this is to give me problems and for me It took “more”. Of course this theory assumes that Dut/Fin work exactly like Saw P does. I have found evidence that supports this and evidence that debunks Saw P working exactly like Fin/Dut. This theory also assumes that an epigentic change cannot occur as an accumulative thing.
awesome post.
sorry if you already answered this. Did you ever have Allopregnanolone and tetrahydrodeoxycorticosterone tested? I think this line of thinking has a lot of potential
Fin/Dut are irreversible competitive inhibitors of 5-AR. Let’s say that once a particular 5-ar enzyme becomes part of the aforementioned covalently-bonded 5-ar+NaDP+Fin/Dut complex, then it is permanently inert.
For the remainder of its existence, the active site is blocked and it will no longer act as a catalyst in the reaction of T + NaDPH -> DHT + NaDP+ and it will eventually be destroyed by the cell’s waste management systems.
Meanwhile, new 5-ar enzymes are being produced and if fin/dut is present in appreciable concentrations, those new enzymes will share the same fate. If no fin/dut is present, they will catalyze the reaction of T + NaDPH -> DHT + NaDP+
More discussion about the mechanism of action of finasteride here:
Not going to go into a lecture about seeing the forest for the trees, but the result of taking fin or dut is a reduction of DHT and some other products of 5-ar metabolism. Either drug could work by sucking 5-ar into another dimension on a molecule-per-molecule basis and the result would still be the same.
I haven’t because there’s no clear path to enhance the 3a-HSD or 3b-HSD complex. It is my understanding that low Allopregnanolone and tetrahydrodeoxycorticosterone with high cortisol is what prevents us from having good sleep and unless we can fix 3a-HSD and 3b-HSD there’s no point confirming that Allopregnanolone is missing.
It costs hundreds of dollars to have those tested, then maybe some supplements can be taken. It would be dose dependent so from my point of view would fix nothing.
Epigenetic modifications can occur for all kinds of reason: a bad long term abusive relationship, pollution in the air, and yes, the very supplements we’re taking to ease our symptoms. There’s no telling if the epigenome will or will not react.
I took fin for 20 years and started having symptoms after 17 years. If I had stopped 10 years earlier, would I have had pfs ? Hard to tell. Than there are those who got pfs from 1 pill. Clearly, epigenetic damage can occur from taking a pill as well as from stopping taking it.
But to me, this is somewhat irrelevant as long as I can deal with the symptoms. The permanent solution, if there is one, is to change our epigenome so it gets as close to a healthy person as it can and for this, I study and use the suggestions given to increase longevity. It turns out aging is more related to excess epigenetic damage than telomere shortening. Anyway, tricks like fasting, intermittent fasting, being in ketosis etc… produce sirtuins to work on the epigenome AND increase the length of the telomeres.
Already I see signs of epigenetic damage supposed to be permanent clearing out: I’ve had freckles on my face from a bad sunburn 28 years ago and dermatologist told me it was permanent. They are now disappearing ! 75% gone already and those are caused by epigenetic damage. I suppose it’s working on the rest on my body as well.
“Finasteride was originally thought to act as a competitive inhibitor with nanomolar affinity for 5α-reductase type 2 (12). More recently, it was found that finasteride acts as a mechanism-based inactivator of this enzyme”
“Subsequent to inhibitor binding, there is hydride transfer from the NADPH cofactor to the Δ1-2-ene double bond of finasteride”
“The intermediate enolate tautomerizes at the enzyme active site to form a bisubstrate analogue in which dihydrofinasteride is covalently bound to NADP+ The bisubstrate analogue has subnanomolar affinity for 5α-reductase type 2”
“Both AKR1D1 and 5α-reductase type 1 play important roles in the hepatic clearance of steroid hormones, suggesting that high dose finasteride may have an adverse effect on hepatic steroid metabolism”
“Inhibition of AKR1D1 by high dose finasteride would also deprive PXR of its natural 5β-pregnane ligands, resulting in diminished CYP3A4 induction”
“By contrast, in addition to being involved in bile acid biosynthesis, 5β-reductase is responsible for generating 5β-pregnanes, which are natural ligands for the pregnane-X receptor (PXR) in the liver”
“PXR is involved in the induction of CYP3A4, which is responsible for the metabolism of a large proportion of drugs (5, 6). Thus both 5α-reductase and 5β-reductase are involved in the formation of potent ligands for nuclear receptors”
“This could result in significant drug-drug interactions. Importantly, finasteride itself is metabolized by CYP3A4, suggesting that high dose finasteride might prevent its own metabolism (27)”
What if (AND THIS IS JUST A THEORY NOT BEING PRESENTED AS FACT) finasteride prevents it’s own metabolizing at least to a degree and if we had issues with PXR and or CYP3A4 that prevented it’s metabolizing and it’s still binding our NADPH. This is just a thought. And if it’s wrong that does not mean it’s smart to move on and not continue to take a closer look at NADPH
again these are just logical ideas and thoughts that may have been overlooked or not looked at close enough
Intersting thoughts on Epigenetic modifications
I have been looking into the point you made about possible low Allopregnanolone and tetrahydrodeoxycorticosterone with possible high cortisol being responsible for our sleep problems. I am in the middle of doing extensive cortisol testing. So far I have had a middle range normal morning and evening plasma cortisol reading. I have had a normal mid range cortisone reading. I have also had a normal cortisol:cortisone ratio test. This tells me that I am producing the right amounts of cortisol and that proper amounts of my cortisol are converting to cortisone. I will also be doing a multiple time per day saliva cortisol test to get the most detailed confirmation possible that my cortisol levels are remaining stable through out the course of the day. So if everything else comes back normal with my cortisol testing I can logically conclude that if I have an issue with cortisol its at that my GC receptors are either down regulated or up regulated. If this ends up being the case my guess is that I am in low in Allopregnanolone and tetrahydrodeoxycorticosterone which caused the GC receptors to become more sensitive to compensate.
There may be no clear path to increasing 3a-HSD or 3b-HSD but that does not mean there is no path at all. I think that Allopregnanolone and tetrahydrodeoxycorticosterone needs to be tested. I’m taking it that far. I am getting it tested. Hundreds of dollars is worth my life.
Cortisol could be high or normal, but the issue is not cortisol or GC in my opinion. Those have not been affected as, IMO, they are not part of the main 5ar complex fin attacked.
3a-HSD neutralizes DHT. If DHT went lower because of 5ari, I believe the body had to lower 3a-HSD to try and maintain some DHT.
Pregnenenolone + 3b-HSD= Progesterone, + 5ar = 5a-DHP, +3a-HSD = Allopregnanolone.
If you’re to test stuff, test progesterone (should be 0.8ng/ml) and you can either test 3b and 3a-HSD, or their metabolite 5a-DHP and Allopregnanolone as the metabolite is more exactly what you want and they indicate if the precursors are working correctly.
If you don’t mind, tell me your results !
ZRTLab.com can do those. If you do it with them, please tell me how much they charge !
I agree with everything you said. The fact that my recent cortisol and cortisone results show that I have proper amounts of active cortisol converting to inactive cortisone confirms that my 11-Beta-Hydroxy Steroid dehydrogenase (11B-HSD) enzymes are working properly. This is important to confirm because your active cortisol could be in proper amounts in plasma but if not enough of it is converting to inactive cortisone this could result in too much cortisol binding to the mineral corticoid receptors brining on similar symptoms that we experience. So ruling this out is good and I did rule this out.
My recent PROGESTERONE tested in @: <0.5 <1.4 ng/mL
So my recent 0.5 Progesterone result is close to your 0.8 Mark. This suggests that my 3B-HSD enzymes are working properly. However, my 3B-Androstanediol (3B-diol) tested in @ below lab reference range low in 2013. Low 3B-diol suggests that my 3B-HSD enzymes are not working correctly because 3B-HSD also converts DHT to 3B-diol. BUT additionally 17B-hydroxysteriod dehydrogenases (17B-HSD) also convert’s DHEA to 3B-diol. So with this being said my low 3B-diol readings may be in result of 17B-hydroxysteriod dehydrogenases (17B-HSD) not working correctly, which, is suggested by the fact that it appears 3B-HSD, is able to convert proper amounts of Pregnenenolone to Progesterone via 3B-HSD.
There is also the possibility that 5AR recovered enough or was not effected enough in certain places to be able to convert proper over all amounts of T-DHT but not in other places where conversion of progesterone into 5a-DHP takes place. Following this logic there is also a possibility that 3a-HSD could be working well enough in certain areas to be able to covert DHT to 3a-diol but not well enough to convert 5a-DHP into Allopregnanolone. Also enzymes such as 3a-Hydroxysteriod dehydrogenase (3a-HSD) can also convert 3a-diol back into DHT and compensate by doing just this when 5AR is no longer working properly. So after really digging into this it becomes clear that the only way for each individual to know their status on each one of these parameters is to get tested for everyone one of them or as many of them as possible including 5a-DHP and Allopregnanolone like you said.
I am looking into as many labs and companies as possible to try to find the best place to have these tested at the best price. I will let you know.