Is this the smoking gun we’ve been looking for? Is this the cause of our issues?
I came across the following info when looking at the side effects of androgen deprivation therapy for prostate cancer.
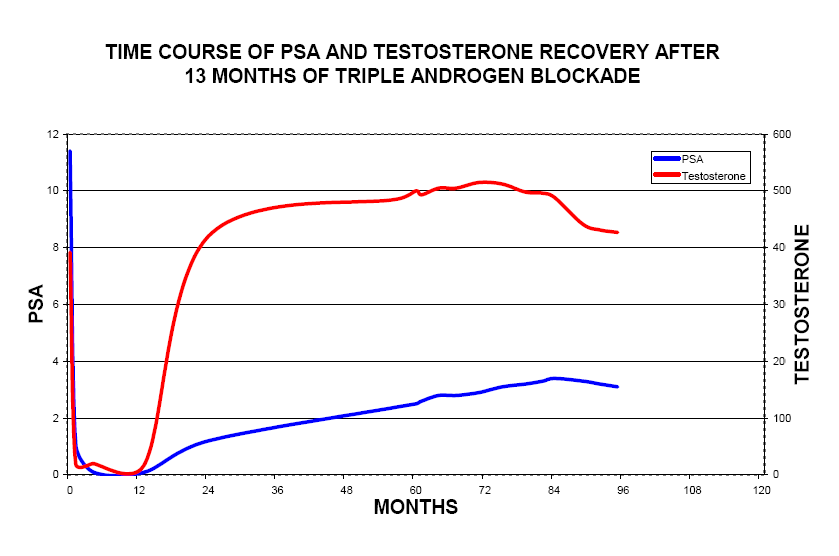
First, notice the attached graph at the BOTTOM of my post, from prostate-cancer.org/educatio … nemia.html
"Figure 5: Serum Testosterone Recovery with Respect to Patient Age and CHB Duration "
[i]Note: CHB means Combined Hormone Blockade. The patients underwent the following:
- (121/133, 91%) received leuprolide acetate (7.5 mg intramuscularly once monthly)
- 116/133 patients (87%) received the antiandrogen flutamide (250 mg orally every 8 hours).
- Forty-eight patients elected to add the 5 alpha-reductase inhibitor finasteride (5 mg orally twice a day) to their CHB. [/i]
… “[Size=4]The persistence of testosterone suppression [/size]in our older patients [Size=4]as long as 24 months off CHB was an unexpected finding[/size]. We have obtained serum LH levels in several of these patients in order to ascertain possible etiologies for this and found them all to be normal or only minimally elevated. This observation is inconsistent with testicular atrophy, where one would expect to see markedly elevated LH levels in response to low serum testosterone. [Size=4]This finding does suggest that prolonged androgen blockade may alter the LHRH receptor[/size], which should respond to low serum testosterone with a secondary rise in levels of LH.”
"Serum testosterone recovered, albeit more gradually than did hemoglobin levels, but was incomplete in patients > 68 years of age who were treated with CHB > 1 year."
The fact that even after discontinuing androgen deprivation therapy, T level recovery was incomplete for those who underwent treatment for greather than 1 year (albeit they were 68+ yrs) is very interesting to note. Also interesting to note is that, if yu look at the graph, they all experienced a “crash” in T levels at 3 months off treatment – similar to how many of us also experienced a post-Fin crash in T levels.
But even more interesting is the fact the researchers have a possible explanation for this: “This finding does suggest that prolonged androgen blockade may alter the LHRH receptor”.
Here’s an even better explanation:
pubmedcentral.nih.gov/articl … id=1831539
“Schally and associates purified the decapeptide gonadotropin-releasing hormone, also referred to as LHRH, in 1971. Chronic exposure to LHRH ultimately suppressed testosterone by desensitizing pituitary cells through downregulation of the LHRH receptors.”
So what is the LHRH receptor? Some background:
prostate-cancer.org/resource/gloss_l.html
LH: luteinizing hormone; a pituitary hormone that stimulates the Leydig cells of the testicles to make the male hormone testosterone
LHRH: luteinizing hormone-releasing hormone (also known as GnRH or gonadotrophin releasing hormone; hormone from the hypothalamus that interacts with the LHRH receptor in the pituitary to release LH). Responsible for stimulating the production of testosterone in the body by interacting with the LHRH receptor to release LH which in turn stimulates cells in the testicles (Leydig cells) to make testosterone.
LHRH analogs (or agonists): Synthetic compounds that are chemically similar to Luteinizing Hormone Releasing Hormone (LHRH), but are sufficiently different that they suppress testicular production of testosterone by binding to the LHRH receptor in the pituitary gland and either have no biological activity and therefore competitively inhibit the action of LHRH, or has LHRH activity that exhausts the production of LH by the pituitary; used in the hormonal treatment of advanced prostate cancer and in the adjuvant and neoadjuvant hormonal treatment of earlier stages of prostate cancer; LHRH agonist (mimics natural LHRH but then shuts down LH production after continuous exposure)
Further to all this – heres is how pituitary desensitization/LHRH receptor changes occur:
acnp.org/g4/GN401000058/CH058.html
Mechanisms of Action of GnRH Agonists
Native GnRH is released from the hypothalamus into the hypothalamo-hypophyseal portal vessels in an episodic (pulse) mode at approximately 90-minute intervals. This pattern of release is associated with pulsatile electrical activity in the hypothalamus. GnRH release leads to episodic release of LH and FSH from the anterior pituitary gland into the systemic circulation after binding to specific GnRH receptors on the cellular surface of gonadotrophs (39).
Receptors occupied by GnRH aggregate into clumps (‘coated pits’), and the receptor-ligand complex is internalized. Cellular responses to GnRH activities of pituitary gonadotrophs are mediated by plasma membrane “second messenger” systems, based on calcium-calmodulin and protein kinase regulatory processes.
In animal models and humans, these physiological events can be disrupted by continuous exposure to native GnRH or administration of long-acting analogs, causing what is termed desensitization to pituitary gonadotrophs. Constant exposure to native GnRH attenuates [decreases] gonadotropin release by downregulating the number of GnRH receptors and/or uncoupling the receptor from distal signal transduction pathways.
The same mechanisms are probably responsible for desensitization induced by GnRH agonistic analogs but may also involve other mechanisms of downregulation, as seen for b-adrenergic receptor in response to excessive agonist stimulation (41,180).
Briefly, when a GnRH agonist is administered, it first leads to supraphysiological LH and FSH secretion and initial stimulation of gonadal function. However, due to the continuous presence of the agonist, increased ligand binding causes maximal physical internalization of agonist-occupied receptors, disrupting gonadotroph receptor replenishment mechanisms and second messenger processes and leads to decreased LH and FSH secretion, with a subsequent reduction in gonadal steroid secretion.
Although pituitary desensitization is probably the principal mechanism responsible for decreased gonadal steroid secretion following GnRH agonist administration, other mechanisms may be involved in this phenomenon (e.g., loss of GnRH pulsatile secretion and subsequent disruption of gonadotropin episodic secretion, changes in serum immunoreactive and bioactive LH concentrations and gonadotropin [LH] gonadal receptor desensitization). Direct inhibitory effects of GnRH agonist at the gonadal level could also play a role.
So what am I getting at here?
Although Finasteride is not an LHRH Agonist, it nontheless does induce an upregulation of LH and Testosterone by 15-20% for as long as the drug is used, in most men. The question is, does Finasteride induce increases in LHRH/GnRH activity/release? Some studies I have read say no, however there are also studies saying that Finasteride has no effect on the hypo-thalamus pituitary axis … claims, which unfortunately, some have found to be untrue after discontinuation.
Since LH release is normally controlled by PULSATILE (aka, non-continous) LHRH (aka GnRH), perhaps continuous use of Finasteride & the resultant constant 15-20% LH/T upregulation led us to have “desensitization to pituitary gonadotrophs”… since we were creating an environment of continous exposure to native GnRH, which “attenuates [decreases] gonadotropin release by downregulating the number of GnRH receptors and/or uncoupling the receptor from distal signal transduction pathways.”
Complicated as that might sound, what I’m basically saying is: perhaps we downregulated and burnt out our pituitary LHRH receptors thanks to Fin’s constant upregulation of Testosterone/LH, so that after we quit, these receptors no longer respond to/recognize low levels of Testosterone in the body as they should (which normally would be to increase LH, to produce more Testosterone). In other words, these receptors/our pituitary has, thanks to Fin, been desensitized and “blinded” to low T, so that they do not think they need to release LHRH to produce more LH, and thus increase T.
… This concept also ties back to the researchers noting the lack of T recovery in the graph (“one would expect to see markedly elevated LH levels in response to low serum testosterone… This finding does suggest that prolonged androgen blockade may alter the LHRH receptor, which should respond to low serum testosterone with a secondary rise in levels of LH”)… for us ex-Fin users, this lack of complete T recovery post-Fin use is eerily similar to those men who did not completely recover their levels after cessation of androgen deprivation therapy.
The question I now have is – is there a way to upregulate LHRH/GnRH receptors, or is it possible to somehow “re-sensitize” the pituitary to “see” low levels of T, in order to increase LHRH/GnRH and produce more LH & T accordingly?
Haven’t done the research, so can’t say yet. And of course, this is all just theoretical conjecture… but at the same time, thought it might potentially offer another possible explanation for some of our post-Fin T-related issues.