
Here’s one Theory that’s out there. Not really a pleasant one. Of course there are many people that take Accutane, so it shouldn’t be as bad as it sounds. (Programmed cell death)
They mention some Retinoic Acid receptor genes to check on.
Apoptosis May Explain the Pharmacological Mode of Action and Adverse Effects of Isotretinoin, Including Teratogenicity
Isotretinoin (13-cis retinoic acid) is the most effective sebum-suppressive drug for the treatment of severe acne. Its effect depends on sebocyte apoptosis, which results from isotretinoin-induced expression of the apoptotic protein tumour necrosis factor-related apoptosis-inducing ligand, insulin-like growth factor-binding protein-3 and neutrophil gelatinase-associated lipocalin. This review proposes that the pharmacological mode of action of isotretinoin in the treatment of severe acne, acute promyelocytic leukaemia, and neuroblastoma results from apoptosis. Furthermore, apoptosis may be the underlying and unifying mechanism of the adverse effects of isotretinoin on neural crest cells (teratogenicity), hippocampal neurones (depression), epidermal keratinocytes and mucosa cells (mucocutaneous side-effects), hair follicle cells (telogen effluvium), intestinal epithelial cells (inflammatory bowel disease), skeletal muscle cells (myalgia and release of creatine kinase), and hepatocytes (release of transaminases and very low-density lipoproteins). Genetic variants of components of the apoptotic signalling cascade, such as RARA polymorphisms, might explain variations in the magnitude of isotretinoin-induced apoptotic signalling and apparently identify subgroups of patients who experience either stronger adverse effects with isotretinoin therapy or resistance to treatment.
INTRODUCTION
Oral isotretinoin (13-cis retinoic acid) is the most effective drug in the treatment of severe nodular recalcitrant acne (1). Isotretinoin was introduced to the market by Hoffman-La Roche in 1982. However, treatment with isotretinoin is associated with adverse effects, the most serious of which is teratogenicity (2, 3). Further manageable adverse effects are mucocutaneous side-effects, increases in transaminases, and hypertriglyceridaemia. Rare adverse effects are depression and inflammatory bowel disease. The aim of this medical hypothesis is to show that the underlying common mechanism that explains the mode of action of isotretinoin and all its adverse effects is apoptosis. The desired apoptotic effect of isotretinoin in the treatment of acne is discussed first, followed by its unwanted adverse effects, including teratogenicity.
SEBOCYTE APOPTOSIS
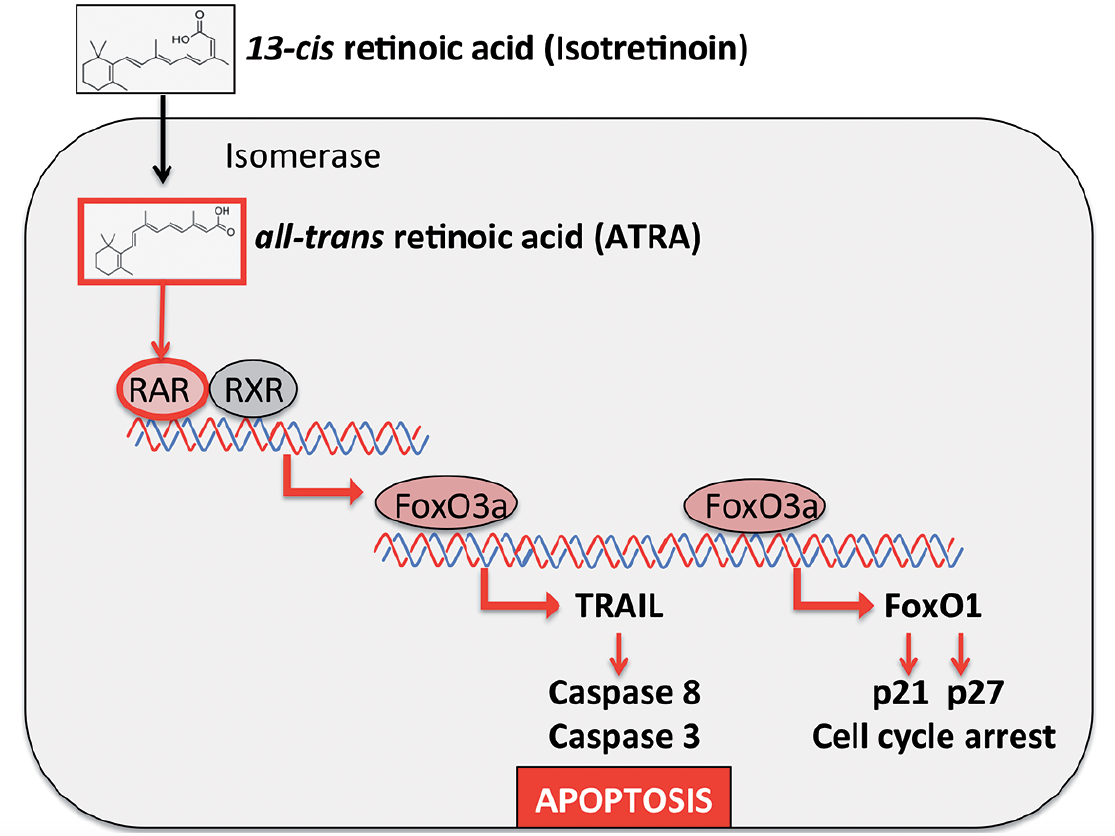
The major pharmacological action of isotretinoin in the treatment of severe acne is sebum suppression (1). Excessive production of sebum and qualitative proinflammatory changes in sebum composition (“acne sebum”) are thought to play a major role in the pathogenesis of acne (4–6). Of all known anti-acne drugs, isotretinoin exhibits the strongest sebum-suppressive effect (7). This sebum-suppressive activity is primarily based on sebocyte apoptosis. Nelson et al. (8) demonstrated that isotretinoin induces apoptosis and cell cycle arrest in human SEB-1 sebocytes. In 2 further studies, Nelson et al. (9) showed that the apoptotic protein tumour necrosis factor-related apoptosis inducing ligand (TRAIL) and neutrophil gelatinase-associated lipocalin (NGAL) contribute to the apoptotic effect of isotretinoin in human sebaceous gland cells (10). Kelhälä et al. (11) recently confirmed increased expression of TRAIL and NGAL in the skin of patients with acne during treatment with isotretinoin. TRAIL (Apo-2L), which induces apoptosis in a number of tumour cell lines, is relatively non-toxic to normal cells. Consistent with its lack of toxicity, TRAIL is constitutively expressed in many human tissues (12). At the promoter level, TRAIL expression is induced by nuclear FoxO transcription factors, especially FoxO3a (13, 14). In patients with acne, FoxO activity is suppressed due to enhanced growth factor signalling (15–17). Insulin-like growth factor-1 (IGF-1) and insulin, which both increase during puberty and under the influence of Western diet (hyperglycaemic carbohydrates and milk) (18, 19), stimulate the kinase Akt, resulting in FoxO phosphorylation, which leads to nuclear FoxO inactivation through cytoplasmic sequestration via 14-3-3 binding (20, 21). FoxOs promote cell growth inhibitory and/or apoptosis signalling, either by stimulating expression of death receptor ligands, such as TRAIL and Fas ligand (FASL), or by inducing the expression of multiple pro-apoptotic members of the Bcl2 family of mitochondria-targeting proteins, or by enhancing levels of cyclin-dependent kinase inhibitors, such as p21 and p27 (13), as demonstrated in isotretinoin-treated SEB-1 sebocytes (8). In sebocytes, the prodrug isotretinoin (13-cis retinoic acid) is isomerized to all-trans retinoic acid (ATRA) ( Fig. 1 ) (22). ATRA upregulates the expression of FoxO3a and TRAIL (23–25). FoxO3a is a key transcription factor of apoptosis (25). In fact, FoxO3a DNA-binding elements exist in the TRAIL promoter, indicating that TRAIL is a direct target of ATRA-induced FoxO3a (14). TRAIL has been shown to be upregulated in human sebaceous gland cells during treatment with isotretinoin (9–11). Thus, it is reasonable to assume that isotretinoin increases FoxO/TRAIL signalling in sebaceous glands of patients with acne, resulting in sebocyte apoptosis. The resulting suppression of sebum, “Kligman’s oil of the acne flame”, is the major anti-acne action of oral isotretinoin therapy.
 Click to show fullsize
Click to show fullsizeFig. 1. Hypothesis of isotretinoin-induced apoptotic signalling explaining the pharmacological and adverse effects of isotretinoin. After isomerization of isotretinoin to all-trans retinoic acid (ATRA) and ATRA binding to retinoic acid receptor (RAR) the transcription factor FoxO3a is upregulated. At the promoter level FoxO3a induces the expression of tumour necrosis factor-related apoptosis inducing ligand (TRAIL) and FoxO1. TRAIL activates the caspase cascade, resulting in apoptosis. FoxO1 mediates cell cycle arrest via upregulation of the cell cycle inhibitors p21 and p27.
FoxOs also play a critical role in the expression of the pro-apoptotic IGF-binding protein-3 (IGFBP3) (26), which acts as a nuclear transcription factor (27). Treatment with isotretinoin significantly increased IGFBP3 in sebocytes (10). Nuclear IGFBP3 interacts with retinoid X receptor-α (RXRα) and retinoic acid receptor-α (RARα) and thereby interferes with the formation of RXR:RAR heterodimers (28). The RXR-IGFBP3 interaction modulates the transcriptional activity of RXRα and is essential for mediating the effects of IGFBP3 on apoptosis (29). FoxO-dependent induction of nuclear IGFBP3 with consecutive activation of RARα may enhance the expression of pro-apoptotic NGAL (30), whose expression at the promoter level is regulated via RARα and RXRα (10). Thus, there is substantial evidence that isotretinoin increases the expression of the pro-apoptotic FoxO proteins, TRAIL, IGFBP3 and NGAL.
There is good reason to assume that pro-apoptotic isotretinoin signalling balances increased Akt-mediated pro-survival signalling in acne skin (31). Increased IGF-1-driven Akt signalling maintains high expression of the anti-apoptotic protein survivin (32), which is upregulated in the serum and skin of patients with acne (133, added in proof). Notably, it has been shown that FoxO3a attenuates survivin expression (33). It is important to note that ATRA-induced FoxO signalling may promote apoptosis, not only in cells of human sebaceous glands and meibomian glands, but also in neural crest cells, neuroblastoma cells, hippocampal neurones, lymphoma cells, melanoma cells, promyelocytic leukaemia cells, and others (30). Thus, the question arises: Are all adverse effects of isotretinoin caused by apoptosis?
APOPTOSIS AND TERMINAL DIFFERENTIATION
Retinoids modify keratinocyte differentiation and, for this reason, are used in the treatment of a variety of disorders of keratinization, including acne. In primary epidermal keratinocytes, ATRA has been demonstrated to regulate many genes associated with cell cycle control and programmed cell death (34). It should be kept in mind that terminal differentiation of epidermal keratinocytes is finally completed by cornification, which is regarded as a specialized process of keratinocyte apoptosis (35, 36). Apoptosis is also involved in the process of holocrine secretion of sebocytes, as well as telogen induction in hair cycling. Intriguingly, the death ligand TRAIL has recently been demonstrated to be involved in keratinocyte differentiation, which requires caspase activation and p63 expression (37). Thus, apoptosis is a critical process for tissue homeostasis and epidermal differentiation. Apoptosis is the result of increased death signalling but decreased pro-survival signalling.
The presented hypothesis, supported by translational evidence, confirms that the undesired effects of isotretinoin on non-target cells are mediated by isotretinoin-induced apoptosis.
APOPTOSIS AND TERATOGENICITY
Treatment with isotretinoin during pregnancy is associated with an increased risk of teratogenicity (38). Preventing foetal exposure to isotretinoin is widely acknowledged as an important safety issue. The iPLEDGE programme is the latest in a series of Food and Drug Administration-mandated risk management programmes designed to prevent pregnancies in female patients of childbearing potential who are taking isotretinoin (39). Isotretinoin exerts teratogenic effects in laboratory animals and humans (40). In humans, a characteristic pattern of malformations involves craniofacial, cardiac, thymic and central nervous system structures (40–43). Some of the most characteristic abnormalities include microtia, anotia, micrognathia, conotruncal heart defects and aortic arch abnormalities, thymic ectopia or aplasia, cerebellar vermis agenesis, and various neuronal migration anomalies (40). The major features of the syndrome are explained by the effect of the drug on neural crest cells. During neural development, massive cell death occurs in vertebrates (44). Programmed cell death (apoptosis) is an evolutionarily conserved contributor to nervous system development and requires a well-regulated balance of apoptotic signalling during most critical periods that ensures appropriate cell differentiation and maturation in the developing nervous system (45). Remarkably, increased neural crest cell apoptosis is also the underlying cause in foetal alcohol syndrome (46). ATRA leads to large-scale reprogramming of cranial neural crest gene expression (47), which results in increased apoptosis (48). Animal studies have confirmed that administration of isotretinoin increases apoptosis of neural crest cells (49–51). Excessive cell death, apparently limited to trigeminal ganglion neuroblasts of placodal origin, follows administration of isotretinoin at the time of ganglion formation and leads to malformations (49). In mouse embryos and primary cell cultures, isotretinoin caused significant overall growth retardation, especially in the primary and secondary palatal processes. In embryos explanted on day 10 of gestation and exposed for 24 or 48 h, the mesenchyme beneath the epithelium of the nasal and maxillary processes contained pyknotic nuclei (signs of apoptosis) as well as dramatically reduced numbers of nuclei incorporating 3H-thymidine (50). Increased neural crest cell apoptosis is the crucial mechanism of isotretinoin-induced craniofacial malformations (52).
Heart defects and aortic arch abnormalities are part of teratogenetic effects of isotretinoin (40–43). Isotretinoin causes a characteristic pattern of heart defects, which result from impaired migration of neural crest cells. Morphogenesis and developmental remodelling of cardiovascular tissues involve coordinated regulation of cell proliferation and apoptosis. In the heart there is clear evidence for focal apoptosis as a contributor to development of the embryonic outflow tract, cardiac valves, conducting system, and the developing coronary vasculature (53). Specific cardiovascular targets of ATRA action include effects on the specification of cardiovascular tissues during early development, such as anteroposterior patterning of the early heart, left/right decisions and cardiac situs and endocardial cushion formation (54–57). Proliferation in heart tissue of whole chick embryo cultures was inhibited in medium with 10–6 M isotretinoin to 62% of the control level in myocardium (58). Taken together, all these observations underline that neural crest cell apoptosis plays the key role in the teratogenicity of isotretinoin.
APOPTOSIS AND DEPRESSION
Recent studies have demonstrated that the hippocampus is one of the brain regions where new neurones are constantly formed; a phenomenon called neurogenesis (59–61). Recent theories for the pathogenesis of depression suggest decreased hippocampal and prefrontal cortex neurogenesis (59–61). In particular, the addition of new neurones within the hippocampus, a limbic region implicated in mood disorders, is compromised in animal models of depression (61). In contrast, antidepressant treatment seems to operate by an increase in neurogenesis, which is chronologically seen during the same period as the clinical improvement (61, 62). Another irregularity in the hippocampus associated with depression is reduction in hippocampal volume. Intriguingly, isotretinoin treatment of mice results in both decreased hippocampal neurogenesis and reduction in hippocampal volume (63, 64). Treatment of hypothalamic cells with 10 ?M isotretinoin for 48 h decreased cell growth to 45.6?±?13% of control (65). Griffin et al. (65) hypothesized that the ability of isotretinoin to decrease hypothalamic cell numbers may contribute to the increased depression-related behaviours observed in mice. A recent study confirmed that intracerebroventricularly applied ATRA to adult rats increased RAR? protein expression in the hippocampus, suggesting an activation of ATRA-induced signalling mechanisms (66). In these rats, ATRA-induced impairments in hippocampal neurogenesis correlated with depression-like symptoms (66). Remarkably, retinoic acid-inducible gene 1 (RAI-1), which increases during neuronal differentiation, was found to be significantly upregulated in brains from patients with schizophrenia, bipolar disorder, or major depression (67). Therefore, isotretinoin-mediated suppression of hippocampal neurogenesis could provide a plausible biological mechanism explaining depressogenic effects of the drug. Individuals with pre-existing impairments of hypothalamic neurogenesis may be at higher risk for the development of isotretinoin-augmented depression. In fact, the literature reviewed from 1960 to June 2010 by Bremner et al. (68) is consistent with isotretinoin-associated depression in a subgroup of vulnerable individuals. The increased risk of isotretinoin-mediated depression is of recent interest in psychiatric research (69–71). Suarez et al. (70) compared isotretinoin-treated vs. non-isotretinoin-treated patients with acne and reported that 13.8% vs. 8.3% of patients developed clinically significant anxiety and/or depression during treatment, respectively. They also concluded that some individuals susceptible to isotretinoin-induced depression do exist, which is in accordance with the author’s own therapeutic experience with oral isotretinoin over more than 30 years. Sundström et al. (72) concluded that an increased risk of attempted suicide was apparent up to 6 months after the end of treatment with isotretinoin. Marron et al. (73) reported a significant reduction in anxiety and depression in isotretinoin-treated patients with acne. However, it is important to realize that acne-mediated depression and isotretinoin-induced depression in a subgroup of patients are 2 different pathogenic entities, which may escape correct detection by epidemiological studies. A subgroup of patients with acne may be more susceptible for isotretinoin-induced neurogenic apoptosis than the great majority of patients who experience an improvement in quality of life after successful acne treatment.
APOPTOSIS AND INFLAMMATORY BOWEL DISEASE
Although the great majority of patients treated with isotretinoin do not experience the development or aggravation of inflammatory bowel disease (IBD) (74–79), an increased risk of IBD in a subgroup of patients cannot be excluded (80–84). It is noteworthy, that a recent study reported excessive death of intestinal epithelial cells (IEC) in the ileal and colonic epithelium to represent a major pathogenetic feature of IBD (85). However, the precise mode of IEC death in IBD has not yet been defined. It is thus conceivable that, in a subgroup of predisposed patients prone to develop IBD, treatment with isotretinoin may aggravate pre-existing IEC apoptosis, thereby promoting the development of IBD, although no scientific evidence is yet available.
APOPTOSIS AND MUCOCUTANEOUS SIDE-EFFECTS
Mucocutaneous adverse effects of oral isotretinoin treatment are dose-dependent and predominantly reflect a decreased production of sebum, reduced thickness of stratum corneum, and altered skin barrier function with increased transepidermal water loss (86). Skin xerosis, especially on exposed skin, and cheilitis are the earliest and the most frequent side-effects that affect almost all treated patients. It should be kept in mind that epidermal keratinocytes undergo a unique form of terminal differentiation and programmed cell death, known as cornification (35, 36). Sebocytes also die in the process of holocrine secretion, which is a less understood process of programmed cell death (35). Notably, ATRA regulates many genes associated with cell cycle control and programmed cell death (34). Genes induced in epidermal keratinocytes by ATRA are the caspases CASP3, CASP6, CASP7 and CASP9 (34). Importantly, ATRA has been shown to induce TRAIL in leukaemia cells (87, 88), which is obviously the therapeutic mode of action of isotretinoin in acute promyelocytic leukaemia and adult T cell leukaemia (89, 90). Wu et al. (37) recently demonstrated that, in keratinocytes, TRAIL is an inducer of both differentiation and apoptosis. Treatment with isotretinoin via activation of TRAIL-induced apoptotic signalling apparently disturbs the homeostatic balance between epidermal cell growth and cell death, which compromises epidermal barrier function (91).
HEPATOCYTE APOPTOSIS
Oral treatment with isotretinoin is often associated with moderate increases in serum levels in alanine aminotransferase (ALT), aspartate aminotransferase (AST), and very low-density lipoproteins (VLDL) triglycerides (92–95). There is good reason to assume that isotretinoin-induced hepatocyte apoptosis explains raised serum transaminase concentrations. Indeed, it has been demonstrated in 2 hepatoma cell lines (Hep3B, HepG2) that ATRA and isotretinoin induce hepatocyte apoptosis (96). However, at present there is no direct evidence that isotretinoin induces hepatocyte apoptosis in treated patients. Upregulated expression of isotretinoin-induced FoxO proteins may explain increased FoxO-mediated expression of hepatic TRAIL (13, 14). In fact, TRAIL has been demonstrated to play a critical role in hepatic cell death and hepatic inflammation (97). Isotretinoin-mediated FoxO1 signalling may also explain isotretinoin-induced hypertriglyceridaemia (30, 98, 99). FoxO1 upregulates microsomal triglyceride transfer protein, which increases hepatic synthesis of triglyceride-rich VLDL (100, 101). Future studies are needed to examine the precise role of upregulated FoxOs in isotretinoin-induced hypertriglyceridaemia.
APOPTOSIS AND MYOPATHY
Muscle-related complaints and increased blood levels of creatine kinase (CK), a specific marker of muscle destruction, have been reported in 16–51% of patients with acne treated with isotretinoin (102–104). Oral isotretinoin-associated adverse muscular effects manifest as myalgia and stiffness and, in rare cases, as true myopathy or rhabdomyolysis. CK has been found to be elevated, occasionally by up to 100 times the normal value, particularly in those patients undergoing vigorous physical exercise (105). Increased TRAIL expression was detected recently in myositis muscle fibres (106). Although direct evidence is missing, it is possible that isotretinoin-induced myalgia may be related to TRAIL-mediated muscle cell apoptosis, explaining CK release during treatment with isotretinoin.
APOPTOSIS AND FERTILITY
Previously, Abali et al. (107) conducted a study about the effect of isotretinoin on ovarian reserve in female Sprague-Dawley rats, which showed a deteriorative ovarian reserve. Notably, the number of ovarian follicles with apoptotic granulosa cells was increased in the experimental groups receiving isotretinoin (107). However, Cinar et al. (108) excluded long-term adverse effects of systemic treatment with isotretinoin on female fertility in humans.
A recent study found no adverse effects of oral isotretinoin in patients with acne on male fertility, given a total dose of 120 mg/kg over a period of 6 months (109). At present, there is no concern about adverse effects of isotretinoin in male fertility (110). Nevertheless, animal studies demonstrated impaired spermatogenesis. In rats, administration of 2?mg/ml/day isotretinoin for 21 days decreased the number of cyclin D1 and E2F-positive cells (111). In adult male gerbils (Gerbillus cheesemani) isotretinoin induced almost complete cessation of spermatogenesis and produced alterations in the cytoplasm of Leydig cells (112). In the adult lizard Podarcis sicula, administration of ATRA impaired spermatogenesis and enhanced testicular germ cell apoptosis (113). These studies point to an increased risk of isotretinoin-induced germ cell apoptosis in these retinoid-susceptible species.
APOPTOSIS AND HAIR LOSS
Long-term use of isotretinoin in higher doses affects hair growth and is associated with increased hair loss and telogen effluvium in susceptible individuals (86, 114, 115). Hair follicles undergo repetitive stages of cell proliferation and programmed cell death. The catagen stage of physiological apoptosis is connected with dynamic changes in morphology and alterations in gene expression (116, 117). ATRA induces premature hair follicle regression, leading to a catagen-like stage in human hair follicles (118). Hair shaft elongation declined significantly already after 2 days in the ATRA-treated group, and approximately 80% of the ATRA-treated hair follicles had prematurely entered catagen-like stage at day 6, compared with 30% in the control group. This corresponded to an upregulation of apoptotic cells in ATRA-treated hair follicles (116).
APOPTOSIS OF CANCER CELLS
Isotretinoin is the preferred retinoid in the chemoprevention of xeroderma pigmentosum and nevoid basal cell carcinoma syndrome (119). Systemic retinoids play an important role in the treatment of cutaneous T-cell lymphoma (120). Isotretinoin mediates apoptosis in Dalton’s lymphoma ascites cells (121). Isotretinoin induced growth inhibition and apoptosis in adult T-cell leukaemia (ATL) cells (90, 122). Furthermore, isotretinoin induced apoptosis in B16F-10 melanoma cells (123). Isotretinoin is used in the treatment of acute promyelocytic leukaemia (124, 125). In promyelocytic leukaemia cells, ATRA-induced expression of TRAIL most likely causes leukaemia cell apoptosis (87). The introduction of isotretinoin in the therapy of neuroblastoma, an aggressive childhood tumour, has significantly improved the prognosis of this malignancy (87, 125–127). Notably, neuroblastoma is derived from the peripheral neural crest (125). There is recent evidence that isotretinoin potentiates the apoptotic effects of other drugs, such as melatonin in neuroblastoma cells (128). Thus, isotretinoin-induced apoptosis apparently targets neural crest cells during embryogenesis as well as neural crest cell-derived neuroblastoma cells. Taken together, these data underline that isotretinoin-induced apoptosis is the fundamental chemopreventive and anti-cancer effect of the drug.
CONCLUSION
There is compelling evidence that the major mode of action of isotretinoin is sebocyte apoptosis. Other cells, which are also highly susceptible for isotretinoin-induced apoptosis, such as neural crest cells or neural crest-derived neuroblastoma cells and leukaemia cells, respond with pronounced apoptosis. We have to keep in mind that isotretinoin (13-cis retinoic acid) is the prodrug for cells that are able to isomerize 13-cis- to all-trans retinoic acid (ATRA). The sebocyte is obviously such a cell, which possesses high isomerase activity for the conversion of isotretinoin to ATRA (22). We know that ATRA binds to retinoic acid receptors and thereby modifies the expression of a multitude of genes, including the upregulation of FoxO3a and TRAIL (23–25). At the promoter level, ATRA-induced FoxO3a activates the expression of TRAIL (13, 14) and FoxO1 (129). Finally, TRAIL activates the caspase cascade and orchestrates the programme of apoptosis, whereas FoxO1 induces cell cycle arrest via upregulation of p21 and p27 (30) (Fig. 1).
Genetic polymorphisms or mutations of components of this death-signalling cascade may explain the observed individual susceptibilities enhancing isotretinoin-mediated apoptosis. Remarkably, genetic polymorphisms in the gene RARA, which encodes RAR?, increase adverse effects of isotretinoin (130). Three-locus haplotype (rs2715554 C/T - rs4890109 G/T - rs9303285 T/C) analysis showed that frequencies of CTG and TTG haplotypes are significantly associated with occurrence of arthralgia, myalgia, nosebleeds and headache in patients treated with isotretinoin. In addition, TCG haplotype was associated with nosebleeds and headache, whereas TTT haplotype was associated with arthralgia and myalgia. Furthermore, levels of AST were increased in patients with the TC genotype of rs2715554 polymorphism, whereas the allele T of rs9303285 was found to be protective against developing depression in patients treated with isotretinoin (130). Thus, genetic variations of critical regulators of isotretinoin-induced apoptotic signalling may explain the increased risk of adverse effects or treatment resistance in subgroups of patients receiving systemic isotretinoin. Notably, activation of caspases, the effectors of the apoptotic machinery, has been detected recently in lymphocytes of patients with depression and anxiety (131). Thus, there is good reason to assume that screening for gene polymorphisms that increase susceptibility for isotretinoin-induced apoptosis and isotretinoin-associated depression may, in the future, be helpful to identify individuals with increased isotretinoin-mediated apoptotic signalling. At present, depression screening using health questionnaires may be an appropriate method to detect susceptible individuals, who should be closely monitored during treatment with isotretinoin (132).
In summary, the underlying mechanism of the mode of action and adverse effects of isotretinoin is proposed to be apoptosis ( Table I ). The magnitude of isotretinoin-induced apoptotic signalling obviously depends on genetic variations, such as RARA polymorphisms. These new insights allow a more balanced and distinguished view on the mode of action of isotretinoin, its risk profile, and explains the enhanced susceptibility for individual apoptosis-related adverse effects in subgroups with genetic variations of isotretinoin-induced apoptotic signalling pathways.