
Very hard to state that something is “statistically significant” if only 16 PFS sufferers participated. The doctor I know would laugh me out of the room if I made such a claim. Actually that was one of the main complaints about all the PFS studies he looked at. Its just very hard to get to a decent sample size if the affected community is so tiny.
Yes, that should have been part of my list as well. In the past, many have speculated that finasteride somehow permanently “nuked” 5ar. RM is clearly not of that opinion:
The molecular mechanism of 5α-R inactivation by finasteride is known since two decades, likely resulting in persistent enzymatic inhibition over time, but this does not fully justify the long-lasting alteration of the neuroactive steroid levels
RM is a well known 5ar specialist (in the context of the brain), so this question is squarely in his field of expertise.
`
2 Likes
We’re not that tiny, we just a) don’t have the funds for larger studies and b) many people are often reluctant to participate or contribute (we’re even seeing that with our simple survey or the 23andMe project)
4 Likes
Any chance there would be a way for non US citizens to get thier hands on it cheap to try when it’s available? I’m not sure when it’ll come to Europe otherwise, I’m kind of banking on this medication as nothing else works for me at least.
There seems to be this misconception that a tiny sample size excludes any “statistical significance”.
An example: If a group of 16 healthy young men had an average T level of 745 ng/dl, with a range of 520-968 ng/dl, and a group of 16 unhealthy young men had an average T level of 374 ng/dl, with a range of 246-610, you bet your ass there would be a “statistically significant” difference between the 2 groups.
1 Like
If the doctor would laugh at you this means that he doesn’t know statistics at all, because the statistical significance was evaluated using a statistical hypothesys test (in this case Pearson’s Chi Square Test) that proved that the results is statistically significant at less than 6/1000, this means that the probability of a casual result is lower than 6/1000 (p=0.006). Usually in studies with small samples it is hard to achieve statistically significant results, so the fact that a strong statistically significance is achieved with a small sample indicates an extremely solid results.
4 Likes
So what happens if the 8 or so PFS guys that had the methylation are spread over 2000 PFS patients.Then it would not be statistically relevant right? You cant know since you have not tested at least 1000. What am I not getting here?
1 Like
I don’t understand what you’re sayin’ sorry.
I’m just readin’ the results that say that the two variables: pfs and high methylation of 5ar type 2 gene have an extremely low probability (less than 6/1000) of being independent. This don’t imply causation. To correctly understand the results is required the basical knowledge of statistics.
You guys should make it impossible to see posts on the forum, until users sign up, login, and take the survey. Even for lurkers.
Just until the goals are met.
Just my opinion
3 Likes
This is actually another shortcoming in RM’s paper: He is focusing too heavily on 5ar2, whereby his previous work has shown neurosteroids to be deregulated which have nothing to do with 5ar2 (red/green areas show all down/upregulated NS found). The problem must additionally also be at either the 3b-HSD or even at the P450scc system level, specifically CYP11A1. Unfortunately there was no assessment of Chol substrate levels either.
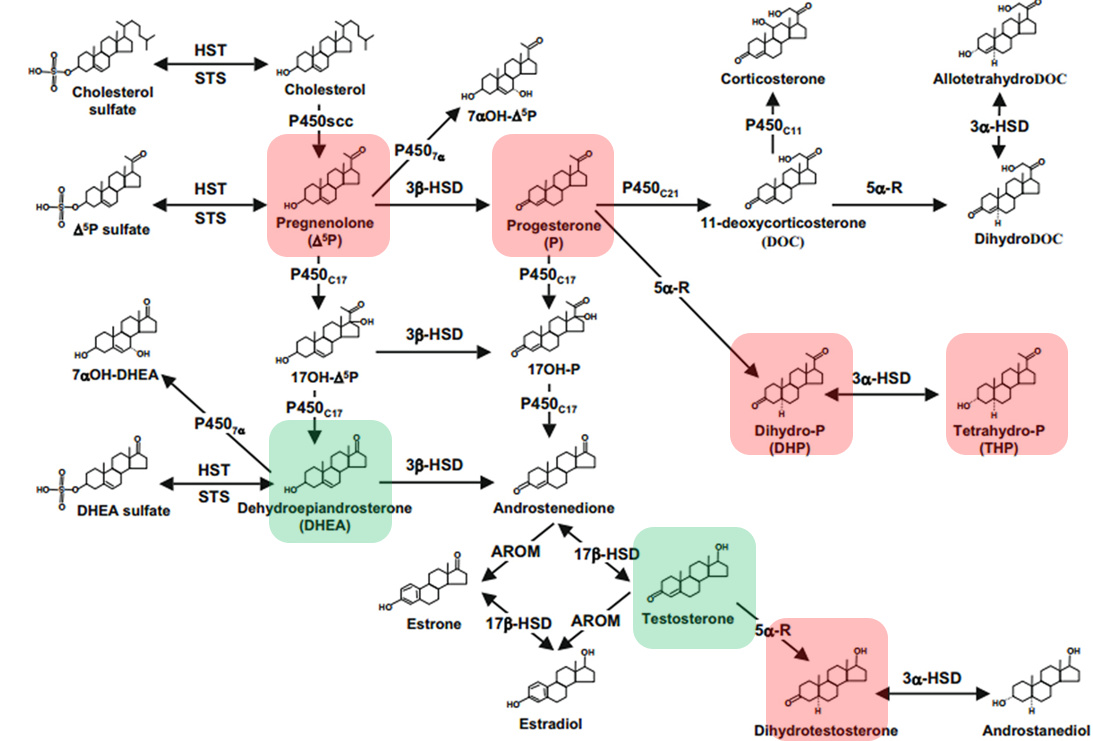
This unfortunately leaves the impression of more shoe-horning on me (like with the symptom profile), which is really a shame, because basically it would otherwise be good work.
4 Likes
But how do you know PFS guys have high methylation of 5ar type 2 gene if you only tested 16?
Statistical inference is used in science to extend observations based on samples to larger populations, it’s impossible to study the whole world’s population for every condition. So there is a nees to produce samples to estimate larger populations. This is how science work, without knowing statistics it is impossible to fully understand the results.
Nice observation! Happy that our science guy noticed this about Melcagni’s studies too.
Would like to mention that HSD3B was one of the most profoundly down-regulated genes in a study of the effect of Accutane on skin cells. Just a hunch that it is indicative of a cell “shutting down” all steroidogenic machinery.
Could you link that study please? Sounds interesting, though things often work differently in the brain than in other sites. Nevertheless, might be something worth replicating with finasteride and AD patients. Certainly easier to do with skin than brain cells, though we would need to pick 5ar2 rich cell lines for finasteride users, unlike what Bhasin (BWH/Harward) did 
1 Like
Suspect you have read this one before. It also shows a decrease in transcription of 5-ar type I and a 3a-HSD enzyme (DHRS9)
A. M. Nelson, W. Zhao, K. L. Gilliland, A. L. Zaenglein, W. Liu, and D. M. Thiboutot, “Temporal changes in gene expression in the skin of patients treated with isotretinoin provide insight into its mechanism of action,” Dermato-Endocrinology , vol. 1, no. 3, pp. 177–187, May 2009.
Full PDF link:
http://www.tandfonline.com/doi/pdf/10.4161/derm.1.3.8258?needAccess=true
…A 6-fold decrease in HSD3B1
I’m not sure. Retinoic acid stimulated neurosteroidogenesis in glial cells and induction of HSD3B was noted, along with p450scc and StAR:
2 Likes
This would be excellent. I’m still somewhat skeptical that it would happen but hey, who knows.
1 Like
I can’t answer that for you. Maybe the company would put out a European product on the market in the future. Or, another company produces an allopreg medication, but I don’t know how patent laws work outside of the USA.
1 Like
8 posts were split to a new topic: Discussion of 5-ar II gene methylation as an effect of AR overexpression
I’m REALLY sorry to interrupt, but seeing as lots of the clever people are all together here I thought I’d ask:
The ONLY mechanism by which DHT is made, is where 5AR converts Testosterone into DHT - is that right?
Thanks all, sorry to butt in.
1 Like
Yes and No. 3a-HSD regulates the amount of DHT available at the receptor (AR) level by converting DHT to Androstenadiol and back. Androstenadiol is a practically inactive androgen. But for that to happen, you need DHT in the first place, which is through conversion of T to DHT via 5ar. Testosterone can be deactivated through 17β-HSD.
Incidentally, scientific research has shown that SSRI class medications such as fluoxetine and paroxetine induce a 63 to 163 fold upregulation of 3α-HSD and 17β-HSDactivity (Griffin LD, 1999) . The upregulation of these androgen reducing enzymes will lead to a significant reduction in DHT at the cellular level. As such, from the point of view of the androgen receptor, the inhibition of 5AR and upgregulation of 3α-HSD/17β-HSD activity have exactly the same effect, namely to reduce the amount of androgens available at the androgen receptor level.
Even though this effect has not yet been investigated outside of the brain, it appears plausible that this is a mechanism which can contribute to causing the same persistent side effects as other androgen depleting substances, given the common persistent side effect profile.
14 Likes